There are 81 treatable inborn errors of metabolism which may account for 5% of all global developmental delay (GDD) / intellectual disability (ID) cases.
There are 91 therapies varying from sick-day management, dietary restrictions & supplements, medications, to haematopoeitic stemcell transplantation. Most are accessible, affordable with acceptable side-effects. These causal treatments may improve or stabilize psychomotor & cognitive development, seizures, neurologic deficits, behavioural/psychiatric disturbances and MRI abnormalities.
Early recognition of one of these treatable conditions in a child with delay or intellectual disability is essential; Time is brain!
This evidenced based Protocol is designed to effectively identify treatable inborn errors of metabolism in patients with global developmental delay / intellectual disability. Please use it to improve health outcomes of affected children in B.C.




NEW TREATMENT & EVINDENCE
The TIDE team has developed and implemented novel treatments for inborn errors of metabolism presenting with intellectual developmental disability, including: the lysine restricted diet and arginine supplementation for Pyridoxine Dependent Epilepsy (ATQ), oral supplementation with creatine, glycine and arginine for Creatine Transporter Deficiency (SLC6A8) as well as the arginine restricted diet and creatine supplementation for GAMT deficiency (GATM).
We have published systematic reviews on evidence of effects and aimed characterize the relevant outcomes, using novel trial methodologies to generate evidence. This individualized approach was used as well to understand individualized outcomes for Phenylketonuria patients treated with a protein restricted diet plus Kuvan. Further studies are in progress.




TIDEX: BIOCHEMICAL PHENOTYPES GUIDING THE DISCOVERY OF INTELLECTUAL DISABILITY GENES
Our TIDEX project aimed to identify novel (potentially treatable) ID genes employing the utility of the metabolic phenotype.
Criteria were applied to select patients for whole exome sequencing (WES): patient with unexplained, Mendelian ID plus metabolic abnormalities. WES was performed for trio’s with customized bio-informatics and subsequent validation.
In 45 families meeting selection criteria, the diagnostic rate was 80%. We discovered 17 new gene defects (various phases of functional validation), including carbonic anhydrase VA deficiency (siblings with hyperammonemia /-lactatemia amenable to treatment with carglumic acid), Rabosyn5 deficiency (early endosomal recycling defect in female with intractable epilepsy), FAAH2 deficiency (in male with adoslecent onset psychiatric disease), SORL1 deficiency (in boy with dementia and gaze palsy); as well as biotin responsive gene and neurotransmitter homeostasis genes.
New phenotypes of known genes were detected in 23, including PIGA deficiency in a boy with MSUD-like brain lesions and progressive multi-organ involvement; AIMP1 deficiency in a girl with neurodegenerative disease and cortical involvement with secondary myelination abnormalities; RMND1 deficiency in a boy with kidney failure, hearing loss, hypotonia and feeding intolerance; and QARS deficiency in a boy with intractable seizures, developmental arrest and supratentorial brain abnormalities.
Our success rate emphasizes the advantages of the biochemical phenotype for gene discovery including: facilitation candidate gene hypothesis, validation causality, and targets for improved management / treatment with direct translation into patient care. We have expanded our study into Omics2TreatID, integrating metabolomics and genomics analyses for gene, pathway and treatment target discovery in 75 or more families.



The goal of the Complex Diagnostic Clinic is to enhance diagnostic success in ‘complex ID’ patients (=patients with global developmental delay / intellectual disability plus prominent other features, in whom the etiology remains unknown) through multispecialty evaluation and communication synergized into a state-of-the-art diagnostic plan using innovative technologies, which is translated into a laymen’s summary for the family.

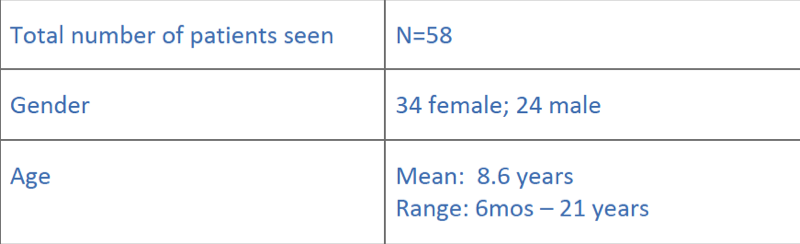
Summary of patients during 17 clinics (2011-2014)
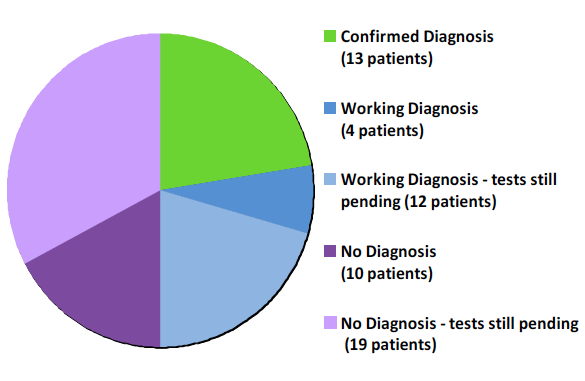
…and their diagnostic yield


The TIDE BC project is located at BC Children’s Hospital in Vancouver, Canada.
TIDE BC is the first Collaborative Area of Innovation funded through the BC Children's Hospital Foundation, Vancouver, Canada.
The TIDE study ran from 2011 - 2019 and is now closed for enrolment.

![]()

![]()
![]()
![]()